Data preparation
繼續(xù)上次的內(nèi)容,下載好數(shù)據(jù)后就可以正式開(kāi)始鑒定了拒课。首先回顧一下狼牺,下載好的數(shù)據(jù)羡儿。
- 基因組序列信息,存儲(chǔ)基因組序列信息的.fasta文件是钥。還有其蛋白質(zhì)序列掠归,也是以.fasta結(jié)尾的文件。一般來(lái)說(shuō)注釋的比較好的基因組都會(huì)含有這些文件悄泥。
- 基因組基因結(jié)構(gòu)注釋信息虏冻。儲(chǔ)存基因的intron,exon弹囚,CDS,gene等坐標(biāo)信息的.gff3或.gtf文件厨相。
- 所感興趣的基因家族隱馬可夫模型,hmm文件
-rw-r----- 1 hhu pawsey0149 9738306 Oct 25 12:24 Arabidopsis_thaliana.TAIR10.41.gff3.gz
-rw-r----- 1 hhu pawsey0149 14623390 Oct 25 12:23 Arabidopsis_thaliana.TAIR10.cds.all.fa.gz
-rw-r----- 1 hhu pawsey0149 36462703 Oct 25 12:23 Arabidopsis_thaliana.TAIR10.dna.toplevel.fa.gz
-rw-r----- 1 hhu pawsey0149 9776319 Oct 25 12:22 Arabidopsis_thaliana.TAIR10.pep.all.fa.gz
-rw-r----- 1 hhu pawsey0149 118379 Oct 25 12:26 NBS-ARC.hmm
基因家族鑒定的工具h(yuǎn)mmer
一般尋找基因家族,都可以通過(guò)保守結(jié)構(gòu)域來(lái)預(yù)測(cè)蛮穿,從而找到物種的某一基因家族庶骄,從而進(jìn)行之后的分析。這里就需要用到HMMER践磅,來(lái)鑒定物種某一基因家族瓢姻。
HMMER3.1下載地址:http://hmmer.org/download.html
HMMER3.1 manual:http://eddylab.org/software/hmmer3/3.1b2/Userguide.pdf
hmmbuild/hmmsearch/hmmscan/hmmalign 這幾個(gè)功能是主要用于蛋白質(zhì)結(jié)構(gòu)與分析和注釋的hmmer中小工具
在鑒定基因家族時(shí),常用到的工具是hmmsearch音诈,里面常用的算法有三種幻碱。一般我們使用--cut_tc算法對(duì)隱馬可夫模型進(jìn)行搜索,tc算法是使用pfam提供的hmm文件中trusted cutoof的值進(jìn)行篩選细溅,相對(duì)比較可靠褥傍。
--cut_ga : use profile's GA gathering cutoffs to set all thresholding
--cut_nc : use profile's NC noise cutoffs to set all thresholding
--cut_tc : use profile's TC trusted cutoffs to set all thresholding
具體實(shí)戰(zhàn)操作
下面會(huì)根據(jù)一篇經(jīng)典文獻(xiàn)中的方法,對(duì)擬南芥進(jìn)行NBS-LRR基因組的探索喇聊。首先恍风,回顧一下文獻(xiàn)看看整體它的思路和方法。
Identification of NBS-LRR genes
Predicted proteins from the cassava genome were scanned using HMMER v3 [39] using the Hidden Markov Model (HMM) corresponding to the Pfam [40] NBS (NB-ARC) family (PF00931; http://pfam.sanger.ac.uk/). From the proteins obtained using the raw NBS HMM, a high-quality protein set (E-value?<?1 × 10?20 and manual verification of an intact NBS domain) was aligned and used to construct a cassava-specific NBS HMM using hmmbuild from the HMMER v3 suite. This new cassava-specific HMM was used, and all proteins with an E-value lower than 0.01 were selected. NBS-LRR genes were further filtered based on manual curation and functional annotation against both the closest homolog from Arabidopsis and the UNIREF100 sequence database. Most of the proteins that were removed had at least a partial kinase domain, but no relationship to NBS-LRR genes; this result was expected because the NBS domain has smaller kinase subdomains
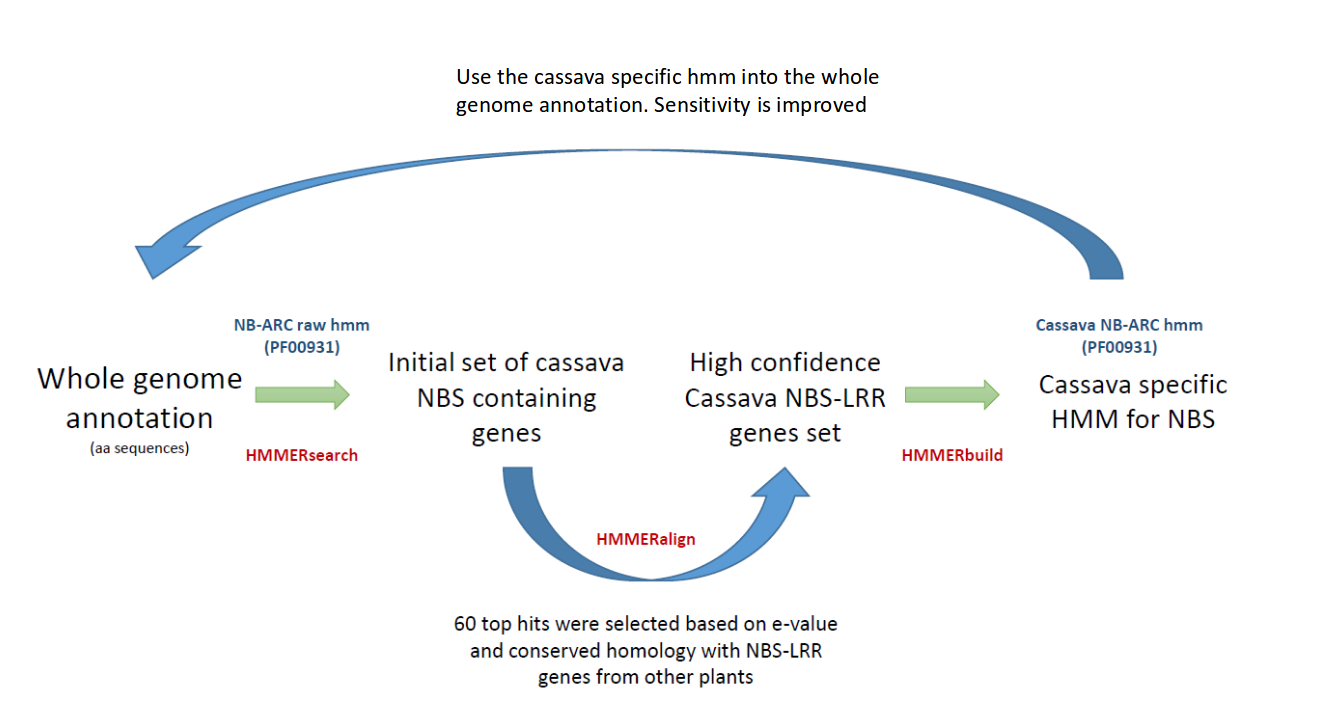
這副圖就是對(duì)應(yīng)了該文章的基因家族鑒定思路誓篱,首先在全基因組的范圍內(nèi)使用hmmersearch和NBS-ARC基因家族的隱馬可夫模型進(jìn)行基因家族的進(jìn)行初步搜索朋贬,接著把質(zhì)量比較高的基因家族候選基因篩選出來(lái)E-value?<?1 × 10?20, 然后使用clustalw2對(duì)高質(zhì)量的序列進(jìn)行多序列比對(duì)窜骄,多序列比對(duì)后锦募,對(duì)這些置信的序列進(jìn)行隱馬可夫模型的構(gòu)建(使用hmmbuild),最后使用該新建的隱馬可夫模型邻遏,進(jìn)一步篩選完整的NSB基因家族序列(需再次過(guò)濾糠亩,找到基因家族的成員數(shù)量一般比第一步初步篩選的多)。
把該流程用到我測(cè)試數(shù)據(jù)中:
##目標(biāo)基因家族搜索
hmmsearch --cut_tc --domtblout NBS-ABC.out NBS-ARC.hmm Arabidopsis_thaliana.TAIR10.pep.all.fa
##簡(jiǎn)單看看運(yùn)算的初步輸出結(jié)果
head NBS-ABC.out
##output
# --- full sequence --- -------------- this domain ------------- hmm coord ali coord env coord
# target name accession tlen query name accession qlen E-value score bias # of c-Evalue i-Evalue score bias from to from to from to acc description of target
#
AT1G61180.1 - 889 NB-ARC PF00931.22 252 1.4e-90 304.3 0.6 1 1 2.2e-92 2.5e-90 303.5 0.6 1 251 156 397 156 398 0.99 pep chromosome:TAIR10:1:22551271:22554684:1 gene:AT1G61180 transcript:AT1G61180.1 gene_biotype:protein_coding transcript_biotype:protein_coding description:LRR and NB-ARC domains-containing disease resistance protein [Source:UniProtKB/TrEMBL;Acc:Q2V4G0]
AT1G61180.2 - 899 NB-ARC PF00931.22 252 1.5e-90 304.2 0.6 1 1 2.2e-92 2.5e-90 303.5 0.6 1 251 156 397 156 398 0.99 pep chromosome:TAIR10:1:22551271:22554684:1 gene:AT1G61180 transcript:AT1G61180.2 gene_biotype:protein_coding transcript_biotype:protein_coding description:LRR and NB-ARC domains-containing disease resistance protein [Source:UniProtKB/TrEMBL;Acc:Q2V4G0]
###對(duì)其e-value進(jìn)行篩選准验,篩選出高質(zhì)量的NBS-LRR蛋白質(zhì)序列赎线。
grep -v "#" NBS-ABC.out|awk '($7 + 0) < 1E-20'|cut -f1 -d " "|sort -u > NBS-ARC_qua_id.txt
~/biosoft/seqtk/seqtk subseq Arabidopsis_thaliana.TAIR10.pep.all.fa NBS-ARC_qua_id.txt >NBS-ARC_qua.fa
對(duì)篩選出來(lái)的序列,使用clustalw2進(jìn)行多序列的比較
clustalw2
**************************************************************
******** CLUSTAL 2.1 Multiple Sequence Alignments ********
**************************************************************
1. Sequence Input From Disc
2. Multiple Alignments
3. Profile / Structure Alignments
4. Phylogenetic trees
S. Execute a system command
H. HELP
X. EXIT (leave program)
Your choice: 1
Sequences should all be in 1 file.
7 formats accepted:
NBRF/PIR, EMBL/SwissProt, Pearson (Fasta), GDE, Clustal, GCG/MSF, RSF.
Enter the name of the sequence file : NBS-ARC_qua.fa
**************************************************************
******** CLUSTAL 2.1 Multiple Sequence Alignments ********
**************************************************************
1. Sequence Input From Disc
2. Multiple Alignments
3. Profile / Structure Alignments
4. Phylogenetic trees
S. Execute a system command
H. HELP
X. EXIT (leave program)
Your choice: 1
Sequences should all be in 1 file.
7 formats accepted:
NBRF/PIR, EMBL/SwissProt, Pearson (Fasta), GDE, Clustal, GCG/MSF, RSF.
Enter the name of the sequence file : new_NBS.aln
對(duì)這些置信的序列進(jìn)行隱馬可夫模型的構(gòu)建(使用hmmbuild),構(gòu)建更加準(zhǔn)確地盡可能預(yù)測(cè)所有的基因家族成員糊饱。
hmmbuild NBS-ARC.second.out NBS-ARC_qua.aln
hmmsearch --cut_tc --domtblout NBS-ARC.second.out NBS-ARC_qua.hmm ../Arabidopsis_thaliana.TAIR10.pep.all.fa
最后對(duì)再次對(duì)這些基因進(jìn)行過(guò)濾與提取
grep -v "#" NBS-ABC.second.out|awk '($7 + 0) < 1E-03' | cut -f1 -d " "|sort -u >final.NBS.list
~/biosoft/seqtk/seqtk subseq Arabidopsis_thaliana.TAIR10.pep.all.fa final.NBS.list >final_NBS-ARC_qua.fa
BLAST-based method
除了使用隱馬可夫模型和hmmer搜索的方法外垂寥,使用同源比對(duì)blast的方法也是鑒定基因家族的其中一種方法。
首先我去了NCBI下載所有植物的存在于Ref-seq(一般認(rèn)為是比較置信的植物基因序列)中的NBS序列
makeblastdb -in ref.nbs.plant.fa -dbtype prot
cat blastp.out |awk '$3>75' |cut -f1 |sort -u > blastp_result_id.list
最后我們還可將上述兩種方法重合的gene id另锋,找出兩種方法共有的基因家族滞项,這樣結(jié)果就比較置信了。
comm -12 blastp_result_id.list hmm_out_id.list > common.list
~/biosoft/seqtk/seqtk subseq Arabidopsis_thaliana.TAIR10.pep.all.fa common.list >final_searh_NBS-ARC_qua.fa
最后可以通過(guò)一些網(wǎng)上的保守結(jié)構(gòu)域搜索網(wǎng)頁(yè)砰蠢,進(jìn)一步對(duì)所找出的結(jié)果進(jìn)行驗(yàn)證:
比如:NCBI CD-Search tool
https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi
Pfam的搜索:
https://pfam.xfam.org/search#tabview=tab1
又或者:InterProScan sequence search
https://www.ebi.ac.uk/interpro/search/sequence-search
這些工具都可再次驗(yàn)證所搜尋的蛋白質(zhì)序列是不是含有基因家族對(duì)應(yīng)的domain蓖扑。在查看保守結(jié)構(gòu)域后唉铜,如果該區(qū)域含有NBS所對(duì)應(yīng)的保守結(jié)構(gòu)域台舱,例如LRR區(qū)域等,該蛋白質(zhì)序列可以保留進(jìn)行后續(xù)的分析。如果在該區(qū)域沒(méi)有找到對(duì)應(yīng)的保守區(qū)域竞惋,為了分析的嚴(yán)謹(jǐn)性柜去,需進(jìn)行進(jìn)一步的排查來(lái)確定是否要去掉該序列。這種情況一般分為兩種情況拆宛,第一種就是注釋無(wú)誤嗓奢,該序列確實(shí)丟失了對(duì)應(yīng)的保守結(jié)構(gòu)域,需要去掉浑厚。第二種情況就是注釋有誤股耽,該序列的結(jié)構(gòu)域可能沒(méi)有被完整的保留下來(lái),這種情況應(yīng)該截取該序列的上下游重新注釋分析钳幅。
