重測序技術(shù)簡介
全基因組重測序(Resequencing)是對已知參考基因組序列的物種進(jìn)行不同個體間的基因組測序,并在此基礎(chǔ)上對個體或群體進(jìn)行差異性分析笆呆。通過全基因組重測序,將不同梯度插入片段(Insert-Size)的測序文庫結(jié)合短序列(Short-Reads)粱挡、雙末端(Paired-End)赠幕,可以找到大量的單核苷酸多態(tài)性位點(SNP)、拷貝數(shù)變異(Copy Number Variation抱怔,CNV)劣坊、插入缺失(InDel,Insertion/Deletion)屈留、結(jié)構(gòu)變異(Structure Variation局冰,SV) 等變異信息,應(yīng)用范圍涉及臨床醫(yī)藥研究灌危、群體遺傳學(xué)研究康二、關(guān)聯(lián)分析、進(jìn)化分析等眾多領(lǐng)域勇蝙。
原理
將特定組織或者細(xì)胞中的DNA進(jìn)行隨機(jī)打碎沫勿,構(gòu)建片段為350bp或者500bp的文庫,通過Illumina Hiseq對文庫進(jìn)行高通量測序,從而獲得某一個個體所有DNA序列的信息产雹。
全基因組數(shù)據(jù)分析的必要條件
- 所測物種的序列是有參考基因組的
- 所測序個體與參考基因組之間遺傳差異性不大 (read 比對不上诫惭,很難找到SNP 等突變信息)
評價測序量的指標(biāo)
測序深度是評價測序量的最重要指標(biāo)。測序深度(Sequencing Depth):測序得到的堿基總量(bp)與基因組大新凇(Genome)的比值夕土。
測序覆蓋比例(Sequencing Coverage),指的是基因組上至少被檢測到1次的區(qū)域瘟判,占整個基因組的比例怨绣。
例如,在某1個樣本測序的項目中拷获,基因組平均測序深度為60X篮撑。基因組大部分區(qū)域的測序深度在60X左右匆瓜,但同時依然有一小部分區(qū)域的測序深度低于3X(極低覆蓋或沒有覆蓋)赢笨。
當(dāng)然,這是理想條件下陕壹。在實際情況下的覆蓋度质欲,會低于理想值。主要是由于GC含量偏好糠馆,基因組完整性,個體差異怎憋,重復(fù)序列影響等又碌。

如下圖,在某1個樣本測序的項目中绊袋,基因組平均測序深度為60X毕匀。基因組大部分區(qū)域的測序深度在60X左右癌别,但同時依然有一小部分區(qū)域的測序深度低于3X(極低覆蓋或沒有覆蓋)皂岔。
當(dāng)然,這是理想條件下展姐。在實際情況下的覆蓋度躁垛,會低于理想值。主要是由于GC含量偏好圾笨,基因組完整性教馆,個體差異,重復(fù)序列影響等擂达。
測序深度與基因組覆蓋度之間是一個正相關(guān)的關(guān)系土铺,測序帶來的錯誤率或假陽性結(jié)果會隨著測序深度的提升而下降。重測序的個體,如果采用的是Paired-End悲敷,當(dāng)測序深度達(dá)到10x時基因組的覆蓋度已接近飽和究恤,基因組覆蓋度和測序錯誤率控制均得以保證。
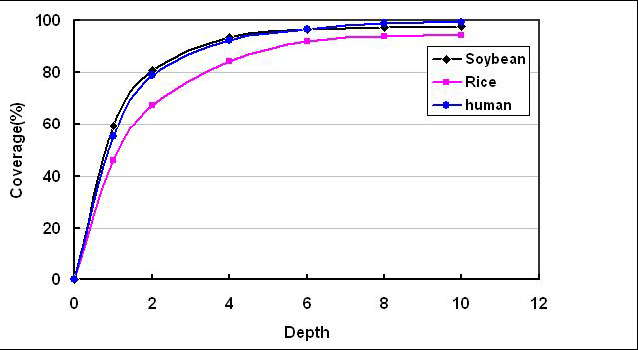
但SNP的檢測率卻沒有達(dá)到飽和后德。這是由于當(dāng)深度達(dá)到10X的時候部宿,雖然基因組大部分區(qū)域已被覆蓋,但在覆蓋到的區(qū)域中探遵,依然有相當(dāng)多的區(qū)域深度小于34X窟赏。SNP檢測的最低深度標(biāo)準(zhǔn)通常為34X。如果沒有達(dá)到這個水準(zhǔn)箱季,則判斷其不可靠涯穷,而在分析結(jié)果中不予接受。為了進(jìn)一步減少低測序深度區(qū)域的比例藏雏,則需要進(jìn)一步提高測序深度拷况。只有測序深度達(dá)到30X的時候,SNP檢測才會達(dá)到飽和

因此掘殴,可以根據(jù)我們的研究目的來選擇相應(yīng)的測序深度赚瘦。

重測序應(yīng)用
目前重測序技術(shù)已廣泛應(yīng)用于農(nóng)學(xué)、醫(yī)學(xué)等各個研究領(lǐng)域奏寨,包括性狀相關(guān)候選基因篩選起意、動植物育種、單基因病篩查病瞳、癌癥篩查等揽咕,快速準(zhǔn)確,對育種和臨床診斷有很好的指導(dǎo)作用套菜。下面舉例一些重測序常用的應(yīng)用范圍和主要思路亲善。
- 個體重測序,突變體檢查 (對每個個體的位點進(jìn)行掃描)
- 混池重測序逗柴,群體進(jìn)化分析 (通過SNP進(jìn)行后續(xù)分析)
- BSA蛹头,遺傳圖譜構(gòu)建
- Hic-主要是做人類比較有用,這里不多說了戏溺。
動植物重測序文章思路
- GWAS與群體進(jìn)化相結(jié)合
隨著GWAS統(tǒng)計方法的不斷完善渣蜗,GWAS能夠適用于大部分物種,將GWAS與群體進(jìn)化結(jié)合分析為性狀關(guān)鍵基因定位提供了一個新的思路于购。
- GWAS與QTL定位相結(jié)合
連鎖分析和關(guān)聯(lián)分析在數(shù)量性狀研究上都具有重要作用袍睡,它們在QTL定位的精度和廣度、提供的信息量肋僧、統(tǒng)計分析方法等方面具有明顯的互補(bǔ)性斑胜。
簡化基因組測序
簡單來說是對與限制性核酸內(nèi)切酶識別位點相關(guān)的DNA進(jìn)行高通量測序控淡。
其實那么多種簡化基因組方法的區(qū)別就在于單酶切還是雙酶切、是否有隨機(jī)打斷止潘、使用不同的內(nèi)切酶掺炭、是否加barcode、接頭設(shè)計等這些細(xì)節(jié)凭戴,本質(zhì)還是一樣的涧狮,就是對基因組進(jìn)行酶切并對酶切片段進(jìn)行測序。在這些方法中么夫,RAD和GBS是使用最廣泛的兩種方法者冤,2b-RAD,dd-RAD档痪,SLAF等都是在這些方法的基礎(chǔ)上在不同細(xì)節(jié)處改良涉枫。
RAD(Restriction site Associated DNA):是與限制性核酸內(nèi)切酶識別位點相關(guān)的DNA。RAD方法對基因組DNA進(jìn)行單酶切腐螟,然后對酶切片段超聲波隨機(jī)打斷愿汰,因此測序得到的read1是位置對齊的,而read2是參差不齊的乐纸,因此可用于denovo聚類拼接衬廷,獲得較長的contig,有利于開發(fā)SSR等分子標(biāo)記汽绢。
GBS(Genotyping-By-Sequencing):是指通過測序進(jìn)行基因分型吗跋。2011年由Elshire, R. J.提出2。GBS方法對基因組DNA進(jìn)行單酶切宁昭,不需要超聲波隨機(jī)打斷小腊,而是利用PCR進(jìn)行片段大小選擇;并且對不同的樣品加上不同的barcode久窟,可對多達(dá)96個樣品進(jìn)行pooling建庫,簡化了建庫步驟本缠,因此比RAD成本更低〕饪福現(xiàn)在的GBS經(jīng)過改良,普遍使用雙酶切了丹锹,雙酶切能夠得到在基因組上分布更均一的酶切片段稀颁。雙酶切的GBS,有時候又被稱為dd-RAD楣黍。
dd-RAD(double-digest RAD匾灶,也可以稱為dd-GBS):是雙酶切的RAD,并且通過切膠來進(jìn)行片段選擇租漂,2012年由Brant K.提出3阶女。其實dd-RAD已經(jīng)放棄了經(jīng)典RAD的超聲波片段化的策略颊糜,dd-RAD的建庫流程和經(jīng)典的GBS更為相似,所以下文我們也將之稱為dd-GBS秃踩。 dd-RAD最大的優(yōu)勢在于衬鱼,由于使用了兩種內(nèi)切酶處理,最終獲得片段在基因組上的分布更加均一憔杨,從而提高了數(shù)據(jù)的有效性鸟赫。由于目前的GBS方法普遍使用雙酶切,并且用電泳切膠來取代PCR擴(kuò)增來選擇片段大小消别,因此dd-RAD幾乎等同于目前的GBS方法了
不同方法之間的比較:

簡化基因組的應(yīng)用
簡化基因組技術(shù)由于降低了基因組的復(fù)雜度抛蚤、比全基因組重測序成本低,因此廣泛應(yīng)用于遺傳圖譜構(gòu)建與QTL定位寻狂、群體進(jìn)化分析岁经、群體遺傳分析、全基因組關(guān)聯(lián)分析等研究領(lǐng)域荆虱。那么蒿偎,這么多種方法,應(yīng)該如何選擇呢怀读?概括來說可以從以下幾方面考慮:
1. 所需標(biāo)記數(shù)
不同的研究目的诉位,所需的標(biāo)記數(shù)量并不完全一樣。通常菜枷,需要在全基因組范圍內(nèi)進(jìn)行功能區(qū)間掃描和功能基因挖掘的研究苍糠,如全基因組關(guān)聯(lián)分析和選擇壓力分析,就需要上萬個高密度的分子標(biāo)記啤誊,而系統(tǒng)發(fā)育關(guān)系岳瞭、地理群體結(jié)構(gòu)、基因流蚊锹、系譜檢測瞳筏、連鎖分析等研究的分子標(biāo)記密度則不需要那么高,一般只需要幾百到幾千個分子標(biāo)記足以完成分析牡昆。
對于基因定位的研究姚炕,不同的研究材料、作圖群體丢烘,也會影響到需要的標(biāo)記數(shù)目柱宦。例如利用自然群體進(jìn)行全基因組關(guān)聯(lián)分析,所需的標(biāo)記數(shù)與物種的LD衰減距離相關(guān)播瞳,物種LD衰減得越快掸刊,所需的標(biāo)記數(shù)就越多。又例如利用作圖群體進(jìn)行連鎖作圖QTL定位赢乓,所需的標(biāo)記數(shù)與作圖群體類型和群體大小有關(guān)忧侧。群體經(jīng)歷的世代越多(如RIL群體)石窑,群體越大,則重組事件越多苍柏,理論上提高標(biāo)記密度可以有效提高遺傳圖譜的質(zhì)量尼斧,所以所需的標(biāo)記數(shù)越多。
因此试吁,可先評估研究所需的標(biāo)記數(shù)棺棵,再選擇適合的簡化基因組技術(shù)。RAD技術(shù)因為對所有酶切位點都檢測熄捍,因此標(biāo)記數(shù)要比GBS多烛恤,適合于需要標(biāo)記密度高的研究,如選擇壓力分析余耽。dd-GBS類的技術(shù)雖然收集的片段偏少缚柏,但標(biāo)記分布更加均一,所以數(shù)據(jù)有效性更高碟贾;并且建庫成本比RAD低币喧,更適合大樣品量的研究。
2.有無參考基因組
如果所研究物種沒有參考基因組袱耽,那么RAD技術(shù)更合適杀餐,因為RAD技術(shù)可以利用不對齊的read2進(jìn)行denovo拼接,再與read1拼接朱巨,可以得到長達(dá)400~500bp的片段史翘,有利于SSR分子標(biāo)記開發(fā)以及后續(xù)的引物設(shè)計。而2b-RAD由于片段過短冀续,容易受重復(fù)序列干擾琼讽,且后期不利于設(shè)計引物驗證測序得到的SNP。因此洪唐,2b-RAD不建議用在沒有參考基因組的物種钻蹬,和大的復(fù)雜的基因組上。
3.研究經(jīng)費
簡化基因組測序與全基因組重測序相比凭需,由于只對酶切片段進(jìn)行測序脉让,因此在測序費用上大大下降。而由于目前的各種簡化基因組技術(shù)都會使用barcode對多個樣品進(jìn)行混合建庫功炮,因此各方法間的建庫成本差異已經(jīng)不大。但RAD文庫構(gòu)建過程中有超聲波打斷步驟术唬、dd-RAD需要使用Pippin Prep等儀器薪伏,因此成本還是要比GBS等高。在實際情況中粗仓,可根據(jù)具體樣品數(shù)和研究經(jīng)費選擇合適的技術(shù)方法嫁怀。
總的來說设捐,RAD和GBS技術(shù)是使用最廣泛的兩種簡化基因組技術(shù),其他的技術(shù)方法都是在這兩者的基礎(chǔ)上的改進(jìn)或細(xì)化塘淑。
基因分型芯片
基因分型芯片:利用已知的SNP位點側(cè)翼的序列設(shè)計探針萝招。探針固定在芯片上后,待測定樣本的DNA與芯片雜交并掃描雜交熒光信號存捺,從而鑒定這些探針位點(SNP位點)的基因型槐沼。最有代表性的品牌是illumina和affymetrix。
與簡化組測序技術(shù)相比較
從以上比較捌治,我們可以認(rèn)為兩種技術(shù)都是高性價比的大規(guī)母诠常基因分型的方法。但最大的不同的是:
- 芯片基因分型本質(zhì)上是對已知SNP多態(tài)位點的掃描肖油,來確定樣本在這個位點的基因型兼吓。(其實很多做人類醫(yī)療測序的都用的是芯片,人類的SNPs多態(tài)性信息已經(jīng)比較完善了)那么森枪,我們需要預(yù)先知道這個物種的基因組SNPs多態(tài)性信息(一般來源大規(guī)模重測序)视搏,然后篩選SNP設(shè)計芯片,才能進(jìn)行后續(xù)的基因分型县袱。打個比方:就是已經(jīng)知道這個位置有個“坑”了浑娜,只是看看坑里到底是沙子還是水。所以芯片只能“分型”显拳,不能“發(fā)現(xiàn)”棚愤。
- 簡化基因組測序本質(zhì)上還是測序,所以哪怕這個物種沒有任何已知的SNPs信息杂数,也能使用簡化基因組測序進(jìn)行檢測宛畦。測序兼顧了“發(fā)現(xiàn)”和“分型”的功能。
兩個技術(shù)的適用范圍
1. 沒有標(biāo)準(zhǔn)化芯片的非模式物種
遇到非主流的無標(biāo)準(zhǔn)化芯片的物種揍移,測序無疑是最佳選擇次和。兼顧了SNP的發(fā)現(xiàn)和基因型分型兩個功能。
2. 有標(biāo)準(zhǔn)化芯片的模式物種
如果你研究的物種是人那伐、豬踏施、牛等這些物種,那么芯片公司的提供的芯片還是不錯的選擇的罕邀。畢竟這些芯片位點都是優(yōu)化過的畅形,基本是比較均勻地覆蓋了相應(yīng)物種的整個基因組。芯片的數(shù)據(jù)相對簡單诉探,后期數(shù)據(jù)的基本處理更簡單日熬。而測序數(shù)據(jù),由于數(shù)據(jù)量大肾胯,后期數(shù)據(jù)的預(yù)處理復(fù)雜且需要較多計算資源竖席。
那么耘纱,模式生物中是否抱定標(biāo)準(zhǔn)化芯片呢?也未必毕荐。主要還是兩點:
1)芯片密度是否滿足你的需求束析?
一些成熟的模式種,例如人憎亚,芯片密度都已經(jīng)達(dá)到了兆級別员寇。但對于某些農(nóng)業(yè)種,芯片密度則還停留在較低的水平虽填。例如:illumina玉米和綿羊的芯片丁恭,都停留在50k的密度水平,很久沒有優(yōu)化了斋日。不過也可以理解他們的邏輯牲览,反正芯片和測序儀都是他們家生產(chǎn)的。芯片密度不夠恶守?測序啊第献。哪怕是使用只對基因組一部分進(jìn)行測序的簡化基因組測序,也可以輕松獲得幾百k數(shù)量級的SNP標(biāo)記兔港。所以庸毫,對于芯片密度不夠的情況下,測序是芯片很好的替代品衫樊。
2)對一些稀有位點的檢測
由于設(shè)計芯片只使用群體中具有普遍性的多態(tài)位點飒赃,即這些位點都是在群體中高頻出現(xiàn)的多態(tài)性位點。所以科侈,對一般人群/種群進(jìn)行普遍性的篩查的時候载佳,沒有太大的問題。但如果臀栈,我們研究的群體十分特殊(如蔫慧,研究材料是比較偏的亞種),或研究目標(biāo)就是低頻甚至罕見位點的時候(癌癥权薯、家族遺傳补枚恪),芯片就無能為力了——因為芯片上就沒有這些位點的探針啊盟蚣。
例如黍析,你研究的是藏豬,那么豬的porcine 60k芯片效果就不會太好屎开。因為芯片上的60k位點都是從常見的品種中篩查得到的橄仍,這些位點在藏豬這樣的特殊亞種中可能多態(tài)性較差。而藏豬群體中普遍的多態(tài)性位點,標(biāo)準(zhǔn)化芯片上卻沒有侮繁。那么,這個時候簡化基因組測序的效果會優(yōu)于芯片如孝。測序嘛宪哩,我測的是序列,管你SNPs稀有不稀有通通一網(wǎng)打盡第晰。
總之锁孟,基因分型芯片和簡化基因組測序,各有優(yōu)缺點茁瘦。在具體項目中品抽,應(yīng)該根據(jù)具體情況做選擇。隨著測序價格不斷降低甜熔,測序的確會不斷侵蝕芯片的市場空間圆恤。但芯片依然有其穩(wěn)定、易標(biāo)準(zhǔn)化腔稀、效率高盆昙、成本容易控制等優(yōu)點,在某些需要標(biāo)準(zhǔn)化的領(lǐng)域(例如:醫(yī)療診斷領(lǐng)域)有巨大的應(yīng)用空間焊虏。
