tags: RNA-seq GATK STAR SNP INDEL
RNA-seq 序列比對(duì)
對(duì) RNA-seq 產(chǎn)出的數(shù)據(jù)進(jìn)行變異檢測(cè)分析盗迟,與常規(guī)重測(cè)序的主要區(qū)別就在序列比對(duì)這一步屎债,因?yàn)?RNA-seq 的數(shù)據(jù)是來(lái)自轉(zhuǎn)錄本的,比對(duì)到參考基因組需要跨越轉(zhuǎn)錄剪切位點(diǎn)稼钩,所以 RNA-seq 進(jìn)行變異檢測(cè)的重點(diǎn)就在于跨剪切位點(diǎn)的精確序列比對(duì)狞换。
有一篇文獻(xiàn) systematic evaluation of spliced alignment programs
for RNA-seq data 對(duì) RNA-seq 數(shù)據(jù)常用的 11 款比對(duì)軟件進(jìn)行了詳細(xì)的比較測(cè)試歧胁,例如 tophat2, STAR 等。 GATK 發(fā)布的 RNA-seq 數(shù)據(jù)變異檢測(cè)最佳實(shí)踐流程用了 STAR 2-pass 這一方法進(jìn)行序列比對(duì)桶现,STAR 發(fā)表的文章至今已被引用 1900 余次躲雅,這款軟件的比對(duì)速度很快,也是 ENCODE 項(xiàng)目的御用比對(duì)軟件骡和。
STAR 2-pass 模式需要進(jìn)行兩次序列比對(duì)相赁,建立兩次參考基因組索引。它的思路是第一次建參考基因組索引之后進(jìn)行初步的序列比對(duì)慰于,根據(jù)初步比對(duì)結(jié)果得到該樣本所有的剪切位點(diǎn)信息钮科,包括參考基因組注釋 GTF 中已知的剪切位點(diǎn)和比對(duì)時(shí)新發(fā)現(xiàn)的剪切位點(diǎn),然后利用第一次比對(duì)得到的剪切位點(diǎn)信息重新對(duì)參考基因組建立索引婆赠,然后進(jìn)行第二次的序列比對(duì)绵脯,這樣可以得到更精確的比對(duì)結(jié)果。
這里使用了一個(gè)測(cè)試數(shù)據(jù)演示流程休里,第一次對(duì)參考基因組建索引:
# star 1-pass index
STAR --runThreadN 8 --runMode genomeGenerate \
--genomeDir ./star_index/ \
--genomeFastaFiles ./genome/chrX.fa \
--sjdbGTFfile ./genes/chrX.gtf
然后進(jìn)行第一次序列比對(duì):
#star 1-pass align
STAR --runThreadN 8 --genomeDir ./star_index/ \
--readFilesIn ./samples/ERR188044_chrX_1.fastq.gz ./samples/ERR188044_chrX_2.fastq.gz \
--readFilesCommand zcat \
--outFileNamePrefix ./star_1pass/ERR188044
之后根據(jù)第一次比對(duì)得到的所有剪切位點(diǎn)蛆挫,重新對(duì)參考基因組建立索引:
# star 2-pass index
STAR --runThreadN 8 --runMode genomeGenerate \
--genomeDir ./star_index_2pass/ \
--genomeFastaFiles ./genome/chrX.fa \
--sjdbFileChrStartEnd ./star_1pass/ERR188044SJ.out.tab
再進(jìn)行 STAR 二次序列比對(duì):
# star 2-pass align
STAR --runThreadN 8 --genomeDir ./star_index_2pass/ \
--readFilesIn ./samples/ERR188044_chrX_1.fastq.gz ./samples/ERR188044_chrX_2.fastq.gz \
--readFilesCommand zcat \
--outFileNamePrefix ./star_2pass/ERR188044
由于后面要用 GATK 進(jìn)行 call 變異,還需要對(duì)比對(duì)結(jié)果 SAM 文件進(jìn)行一些處理妙黍,這些都可以用 picard 來(lái)做悴侵,包括 SAM 頭文件添加 @RG 標(biāo)簽,SAM 文件排序并轉(zhuǎn) BAM 格式废境,然后標(biāo)記 duplicate:
# picard Add read groups, sort, mark duplicates, and create index
java -jar picard.jar AddOrReplaceReadGroups \
I=./star_2pass/ERR188044Aligned.out.sam \
O=./star_2pass/ERR188044_rg_added_sorted.bam \
SO=coordinate \
RGID=ERR188044 \
RGLB=rna \
RGPL=illumina \
RGPU=hiseq \
RGSM=ERR188044
java -jar picard.jar MarkDuplicates \
I=./star_2pass/ERR188044_rg_added_sorted.bam \
O=./star_2pass/ERR188044_dedup.bam \
CREATE_INDEX=true \
VALIDATION_STRINGENCY=SILENT \
M=./star_2pass/ERR188044_dedup.metrics
到此序列比對(duì)就完成了畜挨。
使用 GATK 進(jìn)行變異檢測(cè)
感覺(jué) GATK 里面的工具都很慢(相對(duì)于其他的軟件特別慢!)噩凹,都是單線程在跑巴元,有的雖然可以設(shè)置為多線程但是感覺(jué)沒(méi)啥速度上的提升,所以要有點(diǎn)耐心……
由于 STAR 軟件使用的 MAPQ 標(biāo)準(zhǔn)與 GATK 不同驮宴,而且比對(duì)時(shí)會(huì)有 reads 的片段落到內(nèi)含子區(qū)間逮刨,需要進(jìn)行一步 MAPQ 同步和 reads 剪切,使用 GATK 專為 RNA-seq 應(yīng)用開(kāi)發(fā)的工具 SplitNCigarReads 進(jìn)行操縱堵泽,它會(huì)將落在內(nèi)含子區(qū)間的 reads 片段直接切除修己,并對(duì) MAPQ 進(jìn)行調(diào)整。DNA 測(cè)序的重測(cè)序應(yīng)用中也有序列比對(duì)軟件的 MAPQ 與 GATK 無(wú)法直接對(duì)接的情況迎罗,需要進(jìn)行調(diào)整睬愤。
# samtools faidx chrX.fa
# samtools dict chrX.fa
java -jar GenomeAnalysisTK.jar -T SplitNCigarReads \
-R ./genome/chrX.fa \
-I ./star_2pass/ERR188044_dedup.bam \
-o ./star_2pass/ERR188044_dedup_split.bam \
-rf ReassignOneMappingQuality \
-RMQF 255 \
-RMQT 60 \
-U ALLOW_N_CIGAR_READS
之后就是可選的 Indel Realignment,對(duì)已知的 indel 區(qū)域附近的 reads 重新比對(duì)纹安,可以稍微提高 indel 檢測(cè)的真陽(yáng)性率尤辱,如果時(shí)間緊張也可以不做砂豌,這一步影響很輕微 :)
# 可選步驟 IndelRealign
java -jar GenomeAnalysisTK.jar -T RealignerTargetCreator \
-R ./genome/chrX.fa \
-I ./star_2pass/ERR188044_dedup_split.bam \
-o ./star_2pass/ERR188044_realign_interval.list \
-known Mills_and_1000G_gold_standard.indels.hg19.sites.vcf
java -jar GenomeAnalysisTK.jar -T IndelRealigner \
-R ./genome/chrX.fa \
-I ./star_2pass/ERR188044_dedup_split.bam \
-known Mills_and_1000G_gold_standard.indels.hg19.sites.vcf \
-o ./star_2pass/ERR188044_realign.bam \
-targetIntervals ./star_2pass/ERR188044_realign_interval.list
然后還是可選的 BQSR,這一步操作主要是針對(duì)測(cè)序質(zhì)量不太好的數(shù)據(jù)光督,進(jìn)行堿基質(zhì)量再校準(zhǔn)阳距,如果對(duì)自己的測(cè)序數(shù)據(jù)質(zhì)量足夠自信可以省略,2500 之后 Hiseq 儀器的數(shù)據(jù)質(zhì)量都挺不錯(cuò)的结借,可以根據(jù) FastQC 結(jié)果來(lái)決定筐摘。這一步省了又能節(jié)省時(shí)間 :)
# 可選步驟 BQSR
java -jar GenomeAnalysisTK.jar \
-T BaseRecalibrator \
-R ./genome/chrX.fa \
-I ./star_2pass/ERR188044_realign.bam \
-knownSites 1000G_phase1.snps.high_confidence.hg19.sites.vcf \
-knownSites Mills_and_1000G_gold_standard.indels.hg19.sites.vcf \
-o ./star_2pass/ERR188044_recal_data.table
java -jar GenomeAnalysisTK.jar \
-T PrintReads \
-R ./genome/chrX.fa \
-I ./star_2pass/ERR188044_realign.bam \
-BQSR ./star_2pass/ERR188044_recal_data.table \
-o ./star_2pass/ERR188044_BQSR.bam
比如下面的數(shù)據(jù)就可以放心的省略這兩步了:
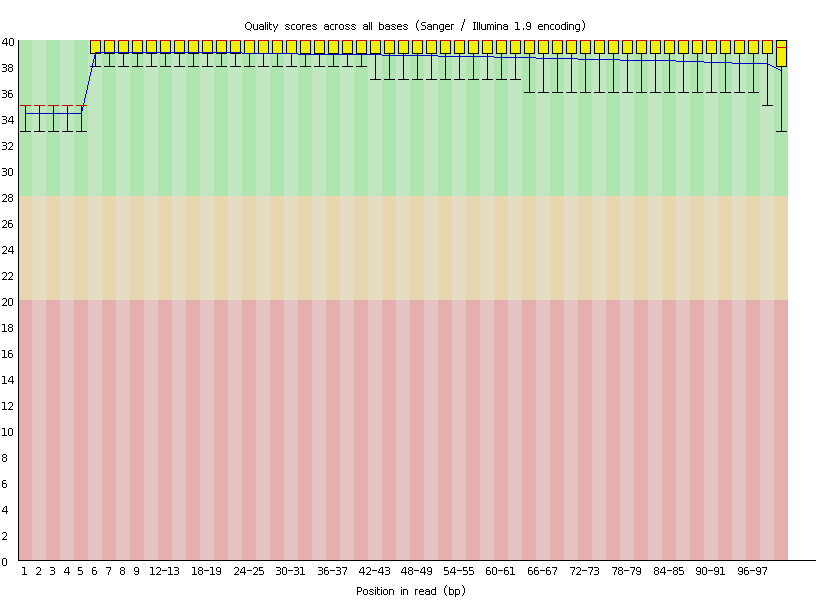
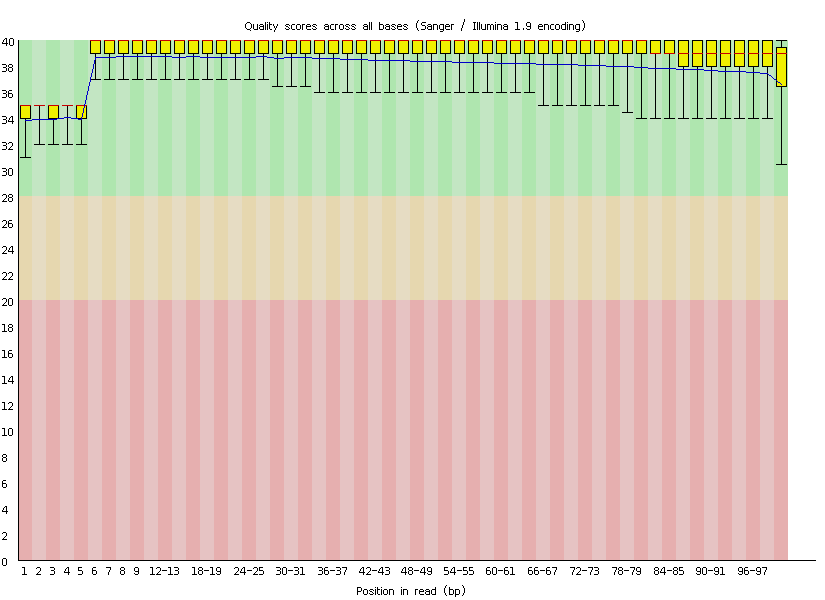
現(xiàn)在終于可以進(jìn)行變異檢測(cè)了,GATK 官網(wǎng)說(shuō) HC 表現(xiàn)比 UC 好船老,所以這里用 HC 進(jìn)行變異檢測(cè):
java -jar GenomeAnalysisTK.jar -T HaplotypeCaller \
-R ./genome/chrX.fa \
-I ./star_2pass/ERR188044_dedup_split.bam \
-dontUseSoftClippedBases \
-stand_call_conf 20.0 \
-o ./star_2pass/ERR188044.vcf
call 完變異之后再進(jìn)行過(guò)濾:
java -jar GenomeAnalysisTK.jar \
-T VariantFiltration \
-R ./genome/chrX.fa \
-V ./star_2pass/ERR188044.vcf \
-window 35 \
-cluster 3 \
-filterName FS -filter "FS > 30.0" \
-filterName QD -filter "QD < 2.0" \
-o ./star_2pass/ERR188044_filtered.vcf
然后就拿到變異檢測(cè)結(jié)果了咖熟,可以用 ANNOVAR 或 SnpEff 或 VEP 進(jìn)行注釋,根據(jù)自己的需要進(jìn)行篩選了努隙。
