當(dāng)一個(gè) NGS 項(xiàng)目中對(duì)同一個(gè)樣本進(jìn)行了 WGS 和 RNA-seq 測(cè)序腰吟,或者對(duì)同一個(gè)腫瘤患者的腫瘤組織和癌旁組織同時(shí)進(jìn)行 WGS / WES / RNA-seq / Chip-seq 測(cè)序,或者由于第一次測(cè)序數(shù)據(jù)量不夠而進(jìn)行加測(cè)得到了兩批數(shù)據(jù),為了排除實(shí)驗(yàn)過程中可能的樣本弄混或者標(biāo)簽貼錯(cuò)节视,就需要驗(yàn)證這不同維度或不同批次的數(shù)據(jù)是否相互匹配。NGSCheckMate 就是這樣一款數(shù)據(jù)質(zhì)控軟件鳄橘,可以滿足以上所有的需求折柠。
為了判斷不同測(cè)序數(shù)據(jù)之間的關(guān)系讳苦,我之前用過 Mendel(Kinship),KING 吩谦,VCFtools (relatedness2 )鸳谜,但是由于軟件接受的輸入數(shù)據(jù)格式問題或者輸出結(jié)果不直觀,都不是很好用式廷,直到我發(fā)現(xiàn)了 NGSCheckMate 咐扭。NGSCheckMate 可以接受 Fastq/BAM/VCF 作為輸入文件,輸出文件包含樣本聚類關(guān)系圖滑废,樣本之間的相關(guān)性系數(shù)蝗肪,樣本是否配對(duì)說明,比之前我用過的其他軟件友好多了蠕趁。
NGSCheckMate 運(yùn)行需要 samtools 在系統(tǒng)路徑中, 安裝之后運(yùn)行也很簡(jiǎn)單穗慕,以 VCF 輸入文件為例:
python ncm.py -V –l INPUT_FILE_list -bed BED_FILE -N test_output –O OUTPUT_DIR
# -l 參數(shù),INPUT_FILE_list 包含所有輸入 VCF 文件的絕對(duì)路徑
# -bed 參數(shù)妻导,BED_FILE 軟件本身自帶逛绵,包括使用的所有 SNP marker 位點(diǎn)
# -N 參數(shù),test_output 輸出文件前綴
# -O 參數(shù)倔韭,OUTPUT_DIR 指定輸出路徑
NGSCheckMate 的文章是 2017 年 3 月份發(fā)表在 Nucleic Acids Research 上的术浪,影響因子 10.162。
摘要
在很多利用 NGS 技術(shù)的研究中寿酌,會(huì)對(duì)同一個(gè)人的不同組織進(jìn)行測(cè)序胰苏,或者對(duì)同一個(gè)組織進(jìn)行不同水平的測(cè)序,例如 DNA-seq醇疼,RNA-seq硕并,Chip-seq。在這種項(xiàng)目中需要添加一個(gè)質(zhì)控環(huán)節(jié)秧荆,來確保不同的數(shù)據(jù)來自同一個(gè)人倔毙。我們開發(fā)了一款簡(jiǎn)潔易用的軟件 NGSCheckMate,利用 SNP 基因型來鑒定數(shù)據(jù)來源乙濒,它可以利用多種格式的數(shù)據(jù)陕赃,如 Fastq,BAM颁股,VCF 來驗(yàn)證不同的數(shù)據(jù)是否來自同一個(gè)人么库。軟件利用已知的單核苷酸多態(tài)性位點(diǎn)(SNP)基因型和等位頻率信息,即使測(cè)序深度不同等位頻率不會(huì)出現(xiàn)太大波動(dòng)甘有,以此來鑒定數(shù)據(jù)是來自同一個(gè)體還是分別來自不同的人诉儒。我們的測(cè)試表明,NGSCheckMate 適用于多種不同水平的數(shù)據(jù)亏掀,包括 WES忱反、WGS运准、RNA-seq、Chip-seq缭受、靶向捕獲測(cè)序和單細(xì)胞全基因組測(cè)序,并且在測(cè)序深度低至 0.5X 時(shí)依然表現(xiàn)良好该互。軟件有免比對(duì)模式米者,可以直接利用 Fastq 原始數(shù)據(jù)快速進(jìn)行數(shù)據(jù)來源鑒定的模塊。我們推薦在 NGS 項(xiàng)目中使用這款工具進(jìn)行數(shù)據(jù)質(zhì)控宇智。軟件可以通過 github 項(xiàng)目主頁獲取蔓搞。
背景介紹
在很多利用 NGS 技術(shù)的研究中,會(huì)比較或者整合來源于同一個(gè)人的多次測(cè)序數(shù)據(jù)随橘,例如對(duì)不同的組織或同一組織不同生理狀態(tài)下的取樣進(jìn)行測(cè)序喂分,比較相同的基因組水平下不同組織間轉(zhuǎn)錄組水平的變化,或者檢測(cè)組織特異性的突變(例如 somatic)机蔗。例如在一些癌癥研究項(xiàng)目中蒲祈,同一個(gè)患者的腫瘤組織和癌旁組織都會(huì)進(jìn)行 WGS 和 RNA-seq 測(cè)序,來檢測(cè)腫瘤中的體細(xì)胞突變及其對(duì)基因表達(dá)水平的影響萝嘁。還有一些情況也需要鑒定數(shù)據(jù)是否來之同一個(gè)體梆掸,例如重復(fù)實(shí)驗(yàn),或者合并不同 lane 上測(cè)得的數(shù)據(jù)(不同 run牙言?)酸钦。
質(zhì)控檢查樣本與標(biāo)簽是否匹配對(duì)于數(shù)據(jù)分析的重要性不言而喻,特別是對(duì)于應(yīng)用于臨床疾病的項(xiàng)目咱枉,患者可能需要根據(jù)數(shù)據(jù)分析的結(jié)果制定治療方案卑硫。盡管我們有標(biāo)準(zhǔn)的實(shí)驗(yàn)質(zhì)控和數(shù)據(jù)質(zhì)控環(huán)節(jié),但是在手工操作環(huán)節(jié)仍有可能出現(xiàn)標(biāo)簽與實(shí)際樣本不匹配的情況蚕断,一旦出現(xiàn)這樣的情況欢伏,要么丟棄這個(gè)可疑樣本的數(shù)據(jù),要么只能重頭開始亿乳。樣本配對(duì)檢測(cè)應(yīng)該作為一個(gè)數(shù)據(jù) QC 環(huán)節(jié)添加進(jìn)流程中颜懊,而且位于流程的越上游越好。
檢測(cè)數(shù)據(jù)是否跟一個(gè)個(gè)體匹配有多種方法风皿,例如微衛(wèi)星序列(STR)檢測(cè)河爹,TCGA 數(shù)據(jù)庫就是利用 PCR 檢測(cè) CODIS 數(shù)據(jù)庫中人群高度多態(tài)性 STR 的方法來驗(yàn)證腫瘤組織和對(duì)應(yīng)的對(duì)照組織是否配對(duì)。但是檢測(cè) STR 的方法對(duì)于 NGS 項(xiàng)目來說并不是很好用桐款,由于大部分 STR 都位于基因組的非編碼區(qū)咸这,因此對(duì)于 RNA-seq 和 WES 測(cè)序不適用,另外很多 STR 序列的長(zhǎng)度都比 NGS 的測(cè)序讀長(zhǎng)要長(zhǎng)魔眨。
還有一種比較普遍的方法媳维,就是利用人群高度多態(tài)性的 SNP 位點(diǎn)來驗(yàn)證兩組數(shù)據(jù)是否配對(duì)酿雪。利用 SNP 位點(diǎn)來驗(yàn)證數(shù)據(jù)已經(jīng)用于 RNA-seq 和 WES 數(shù)據(jù)中。NGS 項(xiàng)目中一個(gè)人的數(shù)據(jù)可以來之不同的 lane侄刽,還有利用 SNP 位點(diǎn)來檢驗(yàn)不同 lane 產(chǎn)出的數(shù)據(jù)是否來之同一個(gè)人的研究指黎。另外,VCFtools 軟件也提供了一個(gè)功能(relatedness2 模塊)州丹,可以利用 VCF 文件檢驗(yàn)兩個(gè)個(gè)體之間的親緣系數(shù)醋安。但是目前這些方法都只適用于特定的數(shù)據(jù)類型,不能用來驗(yàn)證不同類型的數(shù)據(jù)是否來源同一個(gè)人墓毒。
因此吓揪,我們開發(fā)了 NGSCheckMate ,一個(gè)數(shù)據(jù)配對(duì) QC 軟件所计,適用于多種數(shù)據(jù)類型:原始數(shù)據(jù) FASTQ柠辞、比對(duì)數(shù)據(jù) BAM 和變異數(shù)據(jù) VCF。此外 NGSCheckMate 也支持多組學(xué)數(shù)據(jù)之間的配對(duì)檢測(cè)主胧,例如 WES 數(shù)據(jù)和 RNA-seq 數(shù)據(jù)叭首。還有一個(gè)免比對(duì)功能,可以直接利用兩組 FASTQ 數(shù)據(jù)檢測(cè)是否匹配踪栋。NGSCheckMate 可以利用多種類型的數(shù)據(jù)放棒,不同測(cè)序深度的數(shù)據(jù)(> 0.5X),而且運(yùn)行速度非臣河ⅲ快间螟,適用范圍也很廣。
NGSCheckMate 流程和原理
下圖展示了 NGSCheckMate 的基本流程损肛, 輸入文件支持三種格式:Fastq厢破、BAM、VCF治拿,輸出文件也有三個(gè):一個(gè)文本文件摩泪,包含樣本是否配對(duì)檢測(cè)結(jié)果和基因型相關(guān)性系數(shù);一個(gè) PDF 文件劫谅,包含所有樣本聚類關(guān)系圖见坑;一個(gè)樣本聚類關(guān)系 XGMML 文件,可以導(dǎo)入可視化軟件中查看捏检,例如 Cytospace荞驴。
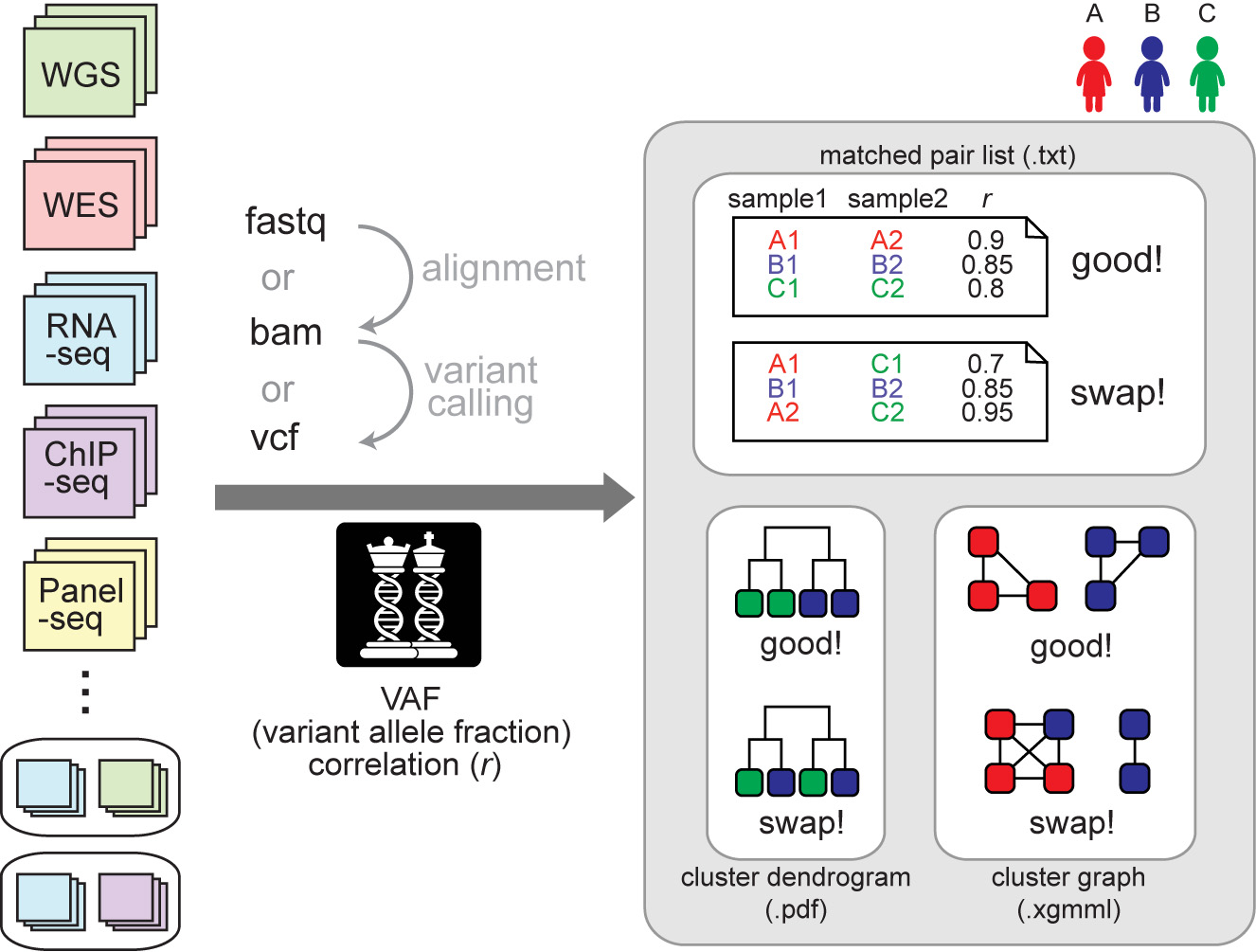
NGSCheckMate 通過計(jì)算兩組輸入數(shù)據(jù)在某些 SNP 位點(diǎn)的 VAF(variant allele fraction) 皮爾森相關(guān)系數(shù)來判斷是否來源同一個(gè)人。
VAF = 支持 ALT 的 reads 數(shù)量 / 覆蓋該 SNP 位點(diǎn)的所有 reads 數(shù)量
下圖展示了 NGSCheckMate 計(jì)算原理贯城。如果輸入的是 BAM 文件熊楼,NGSCheckMate 會(huì)利用 SAMtools mpileup 默認(rèn)參數(shù)來計(jì)算 VAF;如果輸入的是 FASTQ 文件能犯,軟件會(huì)自己搜索覆蓋 SNP 位點(diǎn)的 reads 鲫骗,并統(tǒng)計(jì)支持 ALT 的 reads 數(shù)量和支持 REF 的 reads 數(shù)量犬耻,以此計(jì)算 VAF,當(dāng)然耗時(shí)會(huì)長(zhǎng)些执泰。
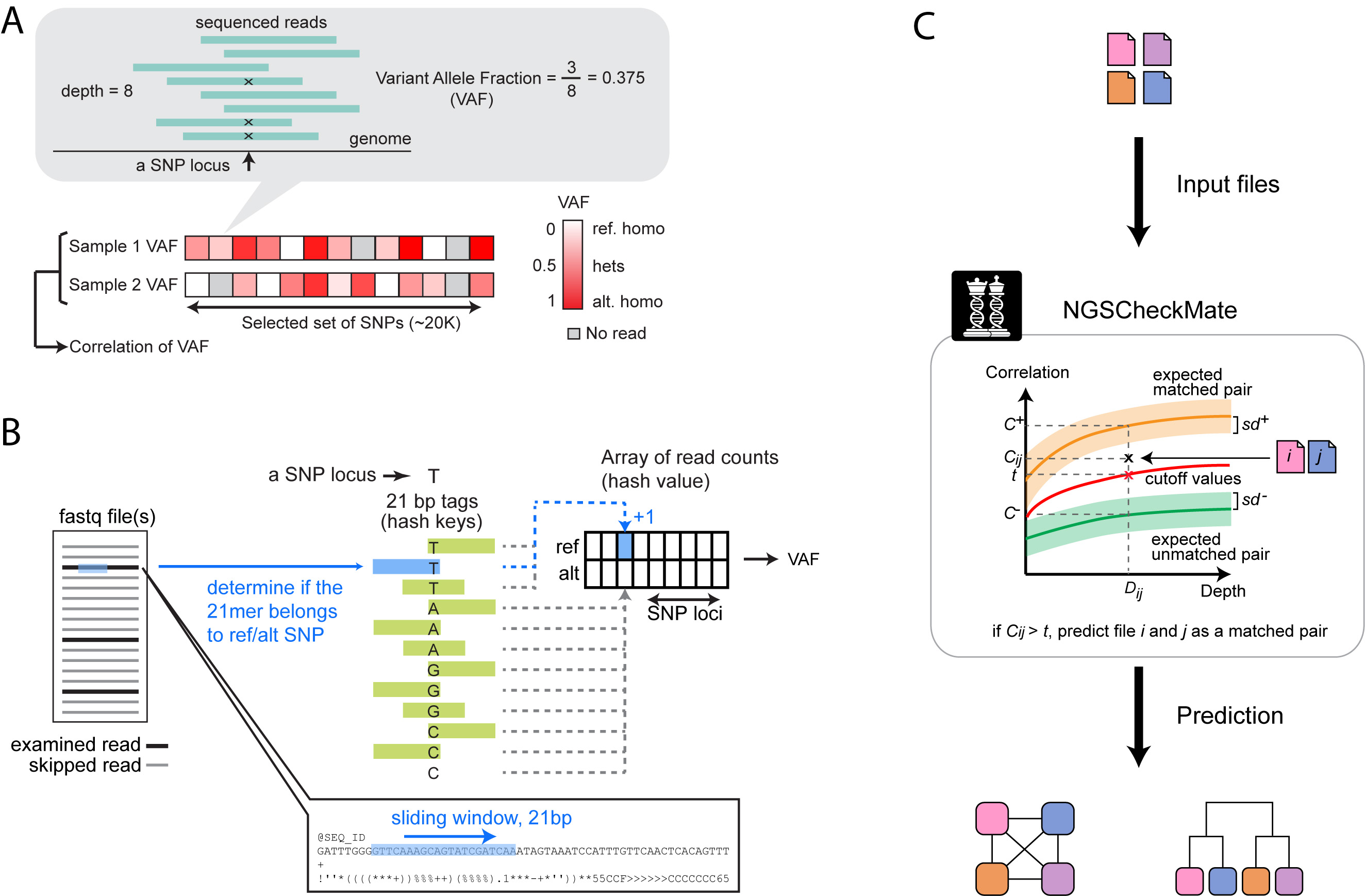
NGSCheckMate 有一個(gè)通過配對(duì)和不配對(duì)的 WGS 數(shù)據(jù)訓(xùn)練好的 VAF 相關(guān)性系數(shù)分布模型枕磁,包括 0.01~60X 的測(cè)序深度下的相關(guān)性系數(shù)分布情況,及配對(duì)和不配對(duì)的相關(guān)性系數(shù)閾值术吝,只需要將輸入數(shù)據(jù)計(jì)算的 VAF 相關(guān)性系數(shù)與模型分布比較即可得出樣本是否匹配的結(jié)論计济。
選擇 SNP 作為鑒別不同個(gè)體的 marker
利用 TCGA 中 40 對(duì) WGS 數(shù)據(jù)測(cè)試了 dbSNP138 的所有 SNP 位點(diǎn)之后,我們選擇了 21067 個(gè)外顯子區(qū)的 SNP 位點(diǎn)作為鑒別不同個(gè)體的 marker 集顿苇。對(duì)于免比對(duì)模式(以 FASTQ 為輸入),是利用其中 11696 個(gè) SNP 位點(diǎn)作為 marker 集税弃。當(dāng)然模擬數(shù)據(jù)顯示纪岁,只使用其中 11696 個(gè) SNP 位點(diǎn)也可以得到與 21067 個(gè) SNP 相同的 VAF 相關(guān)性分布。
免比對(duì)模式
免比對(duì)模式下则果,NGSCheckMate 對(duì) reads 取 21bp k-mer 搜索幔翰,檢測(cè)是否能與 SNP 附近的參考序列比對(duì)上(包括 ALT 和 REF 狀態(tài)),為了確保 21bp k-mer 是唯一比對(duì)的西壮,免比對(duì)模式下把所有參考基因組上下文不唯一的 SNP (SNP 附近的序列在基因組上有多處相同)位點(diǎn)都去掉了遗增。此外為了提高效率,免比對(duì)模式會(huì)從輸入的 FASTQ 中隨機(jī)下采樣款青,保證足夠的準(zhǔn)確率同時(shí)縮短運(yùn)算時(shí)間做修。
構(gòu)建 VAF 相關(guān)性系數(shù)分布模型
我們?cè)?TCGA 數(shù)據(jù)庫中選取了 40 對(duì)的配對(duì)樣本數(shù)據(jù)和非配對(duì)樣本數(shù)據(jù),并對(duì)它們進(jìn)行下采樣(低至 0.01X)抡草,統(tǒng)計(jì)不同測(cè)序深度下的 VAF 相關(guān)性系數(shù)分布情況饰及。得到了一個(gè)非常穩(wěn)健的 VAF 相關(guān)性系數(shù)分布模型(上圖 C 所示),我們就用這個(gè)分布模型來預(yù)測(cè)輸入數(shù)據(jù)是否匹配康震。
對(duì)于輸入的一組數(shù)據(jù)燎含,計(jì)算它們 VAF 相關(guān)系系數(shù),如果落在模型中相同測(cè)序深度下的配對(duì)系數(shù)的分布區(qū)間就預(yù)測(cè)為配對(duì)數(shù)據(jù)腿短,反之亦然屏箍。對(duì)于不同測(cè)序深度數(shù)據(jù),判定是否配對(duì)的 VAF 相關(guān)性系數(shù)閾值也不同橘忱,測(cè)序深度 <1, [1,2), [2,5), [5,10), >=10 閾值分別是 0.38, 0.41, 0.46, 0.55, 0.61 赴魁;但如果樣本都來來自同一個(gè)家庭(父母關(guān)系或兄弟姐妹關(guān)系),閾值會(huì)更嚴(yán)格一些钝诚,分別是 0.50, 0.54, 0.59, 0.69, 0.76 尚粘。如果輸入的兩組數(shù)據(jù)測(cè)序深度不同,則以低測(cè)序深度下的判定結(jié)果為準(zhǔn)敲长。
驗(yàn)證模型的準(zhǔn)確性
從 TCGA 數(shù)據(jù)庫和其他一些研究項(xiàng)目中取 160 對(duì)(腫瘤/對(duì)照) WGS 數(shù)據(jù)秉继,984 對(duì) WES 數(shù)據(jù),170 對(duì) RNA-seq 數(shù)據(jù)泽铛,85 對(duì) panel-seq 數(shù)據(jù),130 對(duì)單細(xì)胞 WGS 數(shù)據(jù)杠茬, 130 組 Chip-seq 數(shù)據(jù)瓢喉,來進(jìn)行測(cè)試栓票。另外,除了 Illumina 平臺(tái)的數(shù)據(jù)逃沿,也測(cè)試了 Ion Torrent 平臺(tái)的兩組數(shù)據(jù)。測(cè)試結(jié)果顯示幻锁,在測(cè)序深度足夠的情況下(>0.5X)感挥,NGSCheckMate 預(yù)測(cè)樣本是否配對(duì)的準(zhǔn)確度接近 100%。
對(duì)于有親緣關(guān)系的個(gè)體之間的 VAF 相關(guān)性系數(shù)分布越败,我們也統(tǒng)計(jì)了 10 個(gè)家庭的 36 份 WES 數(shù)據(jù)触幼,如下圖 A 中綠色/紫色的點(diǎn)。下圖 A 中顯示在測(cè)序深度大于 0.5X 時(shí)究飞,VAF 分布模型可以清晰地將同一個(gè)人的數(shù)據(jù)置谦,有親緣關(guān)系(父母關(guān)系和兄弟姐妹關(guān)系)的數(shù)據(jù),陌生人的數(shù)據(jù)分開亿傅。有親緣關(guān)系的那些數(shù)據(jù)媒峡,父母關(guān)系和兄弟姐妹關(guān)系的數(shù)據(jù)分布幾乎是重疊的(當(dāng)然也無法區(qū)分到底是什么關(guān)系)。
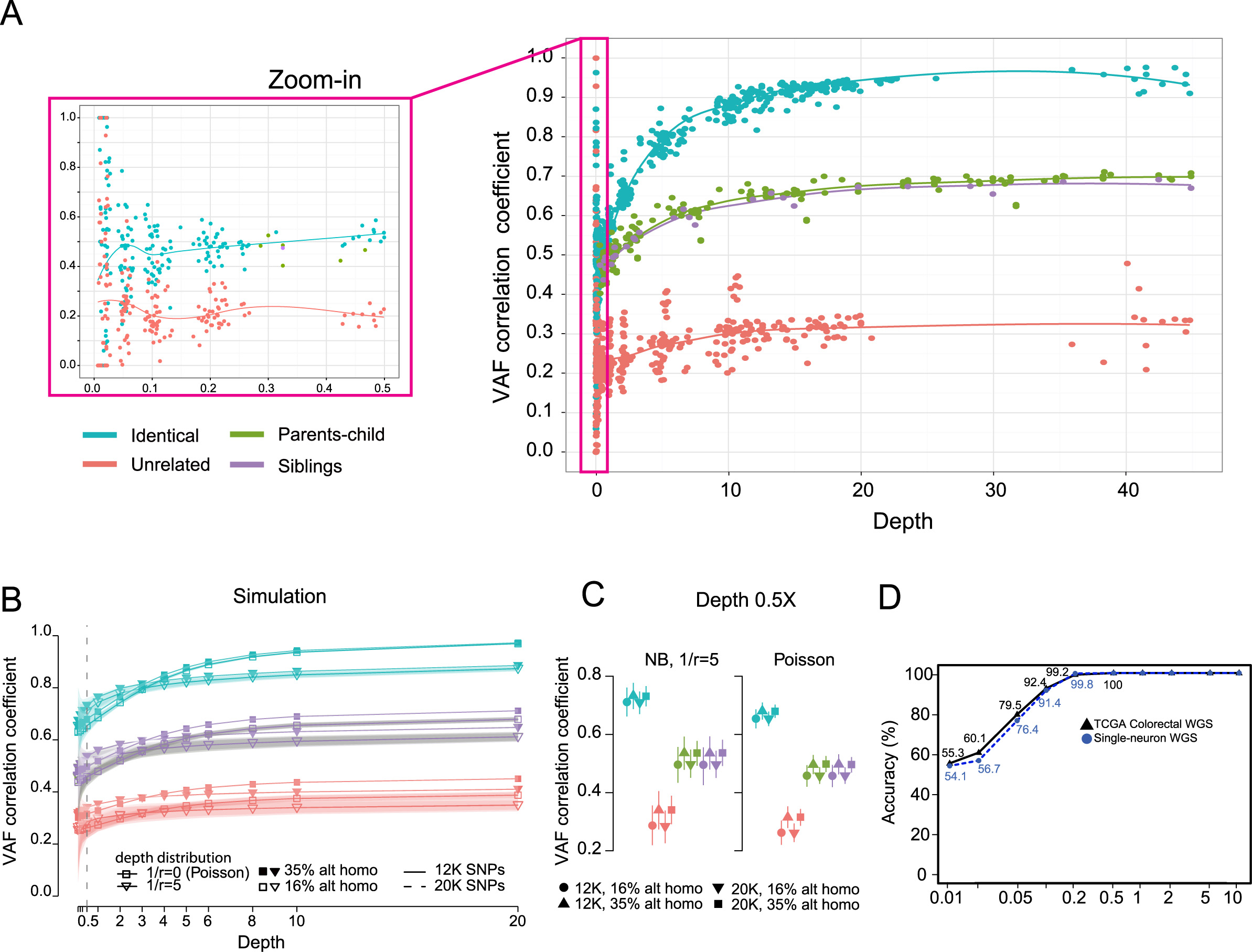
上圖 B 中包含了使用 12K 個(gè) SNP 和使用 20K 個(gè) SNP 計(jì)算的 VAF 相關(guān)性系數(shù)分布葵擎,分布非常接近谅阿。同時(shí)也統(tǒng)計(jì)了 SNP 中純合和雜合的比例對(duì)分布模型的影響,分布也幾乎是重疊的。
上圖 D 中顯示了使用 66 對(duì)(腫瘤/對(duì)照) WGS 數(shù)據(jù)和 36 對(duì)單細(xì)胞 WGS 測(cè)序數(shù)據(jù)進(jìn)行測(cè)試的準(zhǔn)確度签餐,利用下采樣統(tǒng)計(jì)不同測(cè)序深度下的準(zhǔn)確度寓涨。在 0.5X 以上測(cè)序深度時(shí),模型判斷兩組數(shù)據(jù)是否配對(duì)的準(zhǔn)確度接近 100%氯檐。
NGSCheckMate 應(yīng)用實(shí)例:肝癌 WGS 研究項(xiàng)目
在這個(gè)項(xiàng)目中對(duì) 21 個(gè)肝癌患者各自進(jìn)行 3 種組織(腫瘤戒良,癌旁,血液) WGS 測(cè)序冠摄,當(dāng)分析每個(gè)患者的體細(xì)胞突變 SNV 時(shí)候糯崎,我們出乎意料地發(fā)現(xiàn)有不同的患者 somatic SNV 中有大量是相同的,之后進(jìn)一步調(diào)查河泳,發(fā)現(xiàn)可能是部分樣本在操作過程中貼錯(cuò)了標(biāo)簽沃呢,利用 NGSCheckMate 可以鑒定出所有貼錯(cuò)標(biāo)簽的樣本。對(duì)于可能弄混的樣本重新測(cè)序之后拆挥,NGSCheckMate 對(duì)全部 21 個(gè)患者的各自 3 個(gè)樣本給出了正確的聚類薄霜。
此外,利用 NGSCheckMate 也在 TCGA 一項(xiàng)利用低深度 WGS 測(cè)序檢測(cè)胃癌中結(jié)構(gòu)變異的項(xiàng)目中發(fā)現(xiàn) 1 例可能是混樣的數(shù)據(jù)(242 個(gè) BAM 文件的其中一個(gè))竿刁。
NGSCheckMate 運(yùn)行速度和內(nèi)存需求
如果用 VCF 作為輸入文件黄锤,NGSCheckMate 可以在約 3 分鐘之內(nèi)搪缨,利用不超過 200MB 內(nèi)存食拜,檢測(cè) 80 個(gè) WGS 測(cè)序數(shù)據(jù)之間的關(guān)系。運(yùn)算速度很快副编。
如果用 FASTQ 作為輸入文件负甸,NGSCheckMate 為了提高速度,會(huì)進(jìn)行下采樣(取~2X 數(shù)據(jù))痹届,也可以在 11 分鐘左右呻待,利用約 40 MB內(nèi)存,檢測(cè)兩個(gè) WGS 原始數(shù)據(jù)之間的關(guān)系队腐。
總結(jié)
NGSCheckMate 可以非常高效準(zhǔn)確地判定兩組數(shù)據(jù)是否來源與同一個(gè)人蚕捉,適用于多組學(xué)數(shù)據(jù),包括 WGS柴淘,WES迫淹,Chip-seq,RNA-seq为严,panel-seq敛熬,而且由于篩選出來的 20K 個(gè) SNP marker 有足夠的冗余度,只需要測(cè)序深度 >0.5X 就可以給出準(zhǔn)確的判斷結(jié)果第股。NGSCheckMate 的免比對(duì)模式可以在數(shù)據(jù)下機(jī)之后就對(duì) FASTQ 進(jìn)行檢測(cè)应民,盡早發(fā)現(xiàn)可能的樣本與標(biāo)簽弄混的事故。
